АДСОРБЦИЯ
Расстановка ударений: АДСО`РБЦИЯ
АДСОРБЦИЯ (от лат. ad — на, при и sorbeo — поглощаю) — преимущественное концентрирование молекул газа или растворённого в жидкости вещества (адсорбата) на поверхности жидкости или твёрдого тела (адсорбента), а также растворённого в жидкости вещества на границе её раздела с газовой фазой. Частный случай сорбции. Один из важнейших типов поверхностных явлений.
Явление А. связано с тем, что силы межмолекулярного взаимодействия на границе раздела фаз не скомпенсированы, и, следовательно, пограничный слой обладает избытком энергии — свободной поверхностной энергией. В результате притяжения поверхностью раздела фаз находящихся вблизи неё молекул адсорбата свободная поверхностная энергия уменьшается, т. е. процессы А. энергетически выгодны.
В зависимости от характера взаимодействия молекул адсорбата и адсорбента различают физическую А. и хемосорбцию. Физ. А. обусловлена силами межмолекулярного взаимодействия и не сопровождается существ, изменением электронной структуры молекул адсорбата. Физ. А. может быть как монослойной (с образованием мономолекулярного слоя), так и полимолекулярной (многослойной). При А. электролитов из их растворов обычно возникает двойной электрический слой. Если жидкий адсорбат смачивает пористый адсорбент, то в порах последнего может происходить капиллярная конденсация. При физ. А. адсорбир. молекулы обычно обладают поверхностной подвижностью.
При хемосорбции между атомами (молекулами) адсорбента и адсорбата образуется хим. связь, т. о. хемосорбцию можно рассматривать как хим. реакцию, область протекания к-рой ограничена поверхностным слоем. В нек-рых случаях на одной поверхности могут протекать оба типа А. одновременно. В случае не слишком пористых адсорбентов физ. А, имеет место, как правило, при темп-pax ниже критич, темп-ры конденсации адсорбата, хемосорбция же чаще всего протекает при гораздо более высоких темп-рах Однако в нек-рых системах физ. А. может протекать при темп-pax, значительно превышающих критич темп-ру конденсации адсорбата. Как и любые хим реакции, процессы хемосорбции носят специфичный характер (т. е. адсорбент хемосорбирует не любьк молекулы, а лишь те, к-рые вступают в реакцию атомами поверхности); в нек-рых случаях специфич ность может проявляться и при физ. А.
Физ. характеристики А. Количеств, характеристикою А. является величина Г, представляющая собой избыток адсорбата, приходящийся на единицу площади поверхностного слоя, по сравнению с кол-вом адсорбата в единицу объёма фазы адсорбента. Отношение θ=Γ/Γ∞ наз. степенью (или долей) покрытия поверхности (Γ∞ — предельно возможная величина монослойной А. для данной системы).
Процессы А. почти всегда сопровождаются выделением теплоты, наз. теплотой А., к-рая возрастает с увеличением прочности связи адсорбат — адсорбент и составляет обычно 8—25 кДж/моль (иногда до 80 кДж/моль) для физ. А. и, как правило, превышает 80 кДж/моль при хемосорбции. Если хемосорбция сопровождается диссоциацией адсорбир. молекул, может наблюдаться поглощение тепла. По мере заполнения поверхности теплота А. обычно уменьшается в результате неоднородного распределения свободной энергии на поверхности или латерального взаимодействия молекул в адсорбир. слое. Для адсорбентов, обладающих неск. типами адсорбирующих центров (см. ниже), теплота А. может быть различной для разных типов центров, и распределение свободной энергии на поверхности является дискретно-неоднородным. При переходе к полимолекулярной А. теплота А. понижается до величины, близкой к теплоте конденсации адсорбата. Если теплота А. сравнима с поверхностной энергией адсорбента, то в процессе А. может существенно меняться кристаллич. структура поверхности твёрдого тела, причём при физ. А. перестройке подвергаются в осн. поверхности молекулярных кристаллов, а в случае хемосорбции изменение поверхностной структуры наблюдается даже для металлов и ионных кристаллов.
Обратный А. процесс, при к-ром адсорбир. частицы покидают поверхность адсорбента, наз. десорбцией. Десорбция происходит в результате колебат. движения адсорбир. молекул вдоль направления действия силы притяжения между адсорбатом и адсорбентом. Период таких колебаний то обычно составляет 10-13 с. Скорость А. и скорость десорбции могут быть рассчитаны методами статистич. термодинамики. Скорость медленных процессов хемосорбции в большинстве случаев описывается ур-нием
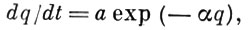
где q — кол-во адсорбир. вещества, а и α — константы, зависящие от темпры. При равенстве скоростей А. и десорбции устанавливается адсорбц. равновесие. Ср. продолжительность времени, к-рое частица находится в адсорбир. состоянии в равновесных условиях (время А.), τ = τ0ехр(Q/RT), где Q — теплота A., R — универсальная газовая постоянная, Т — абс. темп-pa. Принято считать, что А. имеет место в том случае, когда τ достигает величины неск. периодов колебаний адсорбир. молекулы — время, за к-рое между ней и поверхностью успевает установиться энергетич. равновесие. Обычно время физ. А. составляет 10-12—10-6 с, а время хемосорбции — св. 102 с. Время А. служит критерием обратимости процесса А.
Теория А. Единая теория, к-рая описывала бы любые процессы А., пока не создана; существующие частные теоретич. разработки основываются на разл. моделях. Модель локализованной (или центровой) А. предполагает наличие на поверхности адсорбента т. н. центров А., представляющих собой либо строго определ. участки поверхности, на к-рых образуется сильная адсорбц. связь, либо распределённые по поверхности двумерные ячейки со слабым адсорбц. полем (полем сил межмолекулярного взаимодействия). В последнем случае предполагается наличие плотной упаковки молекул адсорбата на поверхности в пределах рассматриваемой ячейки. В основе модели двумерной фазы лежит положение о том, что адсорбир. монослой представляет собой неидеальный двумерный газ, однако полимолекулярное покрытие поверхности адсорбента в данной модели не рассматривается. И, наконец, потенциальная модель А. базируется на представлении о потенц. поле поверхности твёрдого тела, в к-ром адсорбир. газ сжат вблизи поверхности и разрежен в наружных слоях. Эти различные в своей основе модели могут приводить к математически идентичным выражениям, хорошо согласующимся с эксперим. данными. Полуэмпирич. теории, основанные на рассмотренных моделях, не позволяют достаточно строго интерпретировать эксперим. данные, т. к. пока не удаётся учитывать энергетич. неоднородность поверхности, связанную с разл. природой центров А.
Осн. термодинамич. ур-нием, описывающим А., является ур-ние Гиббса:
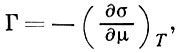
где σ — поверхностное натяжение на границе раздела, μ — химический потенциал адсорбата. Ур-ние Гиббса можно использовать в качестве исходного для вывода ур-ний А. при разл. условиях. К ним, в первую очередь, относятся ур-ния изотерм А., представляющие собой зависимость кол-ва адсорбир. вещества от давления р (или концентрации) адсорбата при пост. темп-ре.
Теория Ленгмюра позволяет вывести ур-ние одной из наиб, простых изотерм А., справедливое при строгой энергетич. однородности поверхности адсорбента, а также при отсутствии на поверхности латерального взаимодействия:
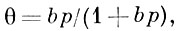
где b — константа, зависящая от темп-ры и характера взаимодействия адсорбат — адсорбент. Типичный вид изотермы Ленгмюра представлен кривой I на рис. 1. При низких значениях р, когда bр<<1 и θ≈bр, изотерма Ленгмюра описывает А. в т. н. области Генри (см. Генри закон).
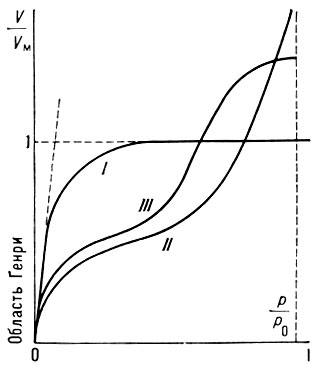
Рис. 1. Наиболее часто вcтречающиеся изотермы адсорбции
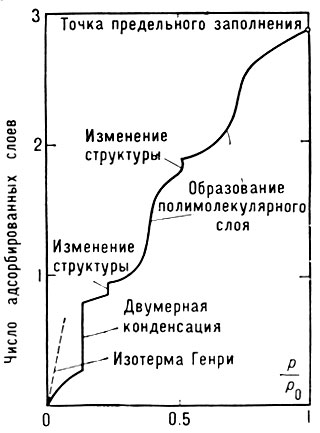
Рис. 2. Обобщённая изотерма изотермы адсорбции (изотерма Холси)
На рис. 1 это отражено прямолинейностью нач. участка изотермы, совпадающего с прямой пунктирной линией. Теория Ленгмюра применима к описанию монослойной физ. А. и хемосорбции, но лишь для огранич. числа систем. Узкая область применимости теории Ленгмюра объясняется, по-видимому, энергетич. неоднородностью поверхности, а также латеральным взаимодействием. Последний фактор в наиб. простом приближении можно учесть путём введения в ур-ние Ленгмюра вместо константы b константу
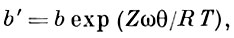 ,
,где Z — координационное число центров А. на поверхности, ω — энергия латерального взаимодействия двух адсорбир. молекул.
Модель Ленгмюра имеет достаточно общий характер и служит основой для построения более развитых теорий, особенно хорошо описывающих хемосорбцию. Так, если допустить, что распределение числа центров А. по энергии носит экспоненц. характер, можно получить ур-ние изотермы Фрейндлиха, в большей степени, чем ур-ние Ленгмюра, применимое для описания процессов не только хемосорбции, но и физ. А.: θ=kp1/n, где n>1 и k — постоянные. Использование экспериментально полученной линейной зависимости теплоты А. от степени заполнения поверхности при ср. значениях последней приводит к изотерме Шлыгина-Фрумкина для хемосорбции: θ=alnbр (а и b — константы).
Вид наиб. часто встречающихся эксперим. типов изотерм (кривые II и III на рис. 1) можно объяснить только на основе теорий, учитывающих полимолекулярность физ. А. Из них наиб. часто применяемой является теория Брунауэра-Эмметта-Теллера (БЭТ), основанная на локализованной модели А. с центрами в виде двумерных ячеек и отсутствии латерального взаимодействия. Её гл. положения — непостоянство толщины адсорбц. слоя на разных участках поверхности и равенство теплот А. теплоте конденсации адсорбата во всех слоях, начиная со второго. Ур-ние изотермы БЭТ имеет вид
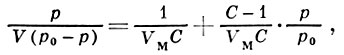
где р0 — давление насыщенного пара адсорбата, V-объём адсорбир. вещества, VM — ёмкость монослоя, C=gexp[(Q — QL/RT], g — статистич. множитель (обычно g ≈1), QL — теплота конденсации адсорбата. При малых относит. давлениях р/р0<<1 ур-ние БЭТ переходит в ур-ние Ленгмюра V/VM =θ=bp/(1+bp) (где b=с/р0). Существуют модификации теории, применимые к пористым адсорбентам в области капиллярной конденсации (кривая III). Теория БЭТ не учитывает латерального взаимодействия, что является её существ, недостатком, наряду с предположением о равенстве теплоты А. теплоте конденсации уже во втором слое. На основе теории БЭТ получено большое число эмпирич. ур-ний, позволяющих описать вид изотерм в нек-рых конкретных адсорбц. системах, но не являющихся универсальными.
В потенц. теории А. (т. н. теория Поляни) полагается, что А. протекает под действием не зависящего от темп-ры потенциала ε(r), численно равного работе, совершаемой адсорбц. силами при переносе молекулы адсорбата из газовой фазы в данную точку, находящуюся на расстоянии r от поверхности адсорбента; при этом свободная энергия адсорбата увеличивается за счёт сжатия последнего и 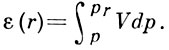 На основании потенц. модели можно для каждой адсорбц. системы построить характеристическую кривую ε=ε(V/VM) [в полимолекулярной области ε=ε(θ) и характеристическая кривая может описывать энергеич. неоднородность поверхности]; с её помощью можно рассчитывать изотермы А. при разл. темп-рах, а также по изотерме А. одного адсорбата рассчитать изотерму А. для другого.
На основании потенц. модели можно для каждой адсорбц. системы построить характеристическую кривую ε=ε(V/VM) [в полимолекулярной области ε=ε(θ) и характеристическая кривая может описывать энергеич. неоднородность поверхности]; с её помощью можно рассчитывать изотермы А. при разл. темп-рах, а также по изотерме А. одного адсорбата рассчитать изотерму А. для другого.
В теории, основанной на модели двумерной фазы, вводят ур-ния состояния двумерного газа, аналогичные соответствующим ур-ниям состояния газа в трёхмерном пространстве, напр, ур-ние состояния типа ур-ния Менделеева-Клапейрона: ηS=nRT, где η — давление в двумерном слое, S — площадь поверхности, занятой адсорбатом, n — число молей адсорбир. вещества. На практике используют обычно одно из ур-ний состояния реального газа и с его помощью выводят ур-ия, описывающие изотермы, аналогичные изотерме I на рис. 1. Кроме того, теория А. на основе модели двумерной фазы находится в определ. соответствии с потенц. моделью, если ф-ция ε(R) имеет вид прямоуг. потенц. ямы.
Плавная форма изотерм А., по-видимому, является следствием энергетич. неоднородности поверхности. В то же время адсорбаты и их комплексы с адсорбентами могут претерпевать на поверхности фазовые переходы, проявляющиеся лишь в условиях строгой энергетич. однородности поверхности в форме ступенек и изломов на эксперим. изотермах. Обобщённая (модельная) изотерма Холси (рис. 2) отражает разл. типы фазовых переходов, соответствующих как субмонослойной области, так и области полимолекулярной А. Возможность всех подобных типов переходов была подтверждена экспериментально.
Все перечисленные модели и теории относятся, в первую очередь, к А. на твёрдых адсорбентах из газовой фазы, однако с небольшими изменениями они пригодны и для описания А. из растворов.
Особое место занимает А. растворённого вещества на границе раздела жидкость — воздух. Согласно ур-нию Гиббса, величина А. таких веществ
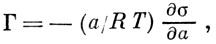 ,
,где а — активность растворённого вещества. Соединения, для к-рых ∂σ/∂a<0, т.е. Г>0, наз. поверхностноактивными веществами (ПАВ); они характеризуются, как правило, полярностью более низкой, чем полярность растворителя. А. ПАВ носит обычно характер монослойной физ. А. и хорошо описывается теорией Ленгмюра.
Помимо изотерм А., на практике часто пользуются изостерами А., выражающими зависимость между равновесным давлением и темп-рой А. для определ. кол-ва адсорбир. вещества. При помощи изостер, полученных методом термодесорбции, осуществляют обычно вычисление теплот А., к-рые можно определять также методом калориметрии. Для изучения А. в настоящее время применяется также разнообразный арсенал совр. методов исследования вещества. Для определения кол-ва адсорбир. вещества, числа адсорбц. центров и величины адсорбир. поверхности используют машинные методы анализа эксперим. изотерм, а также гравиметрич. и радиоизотопный методы и высокотемпературную газовую хроматографию. Поверхность адсорбентов исследуют с помощью методов рентгеновского структурного анализа и электронографии, оже-спектроскопии, мёссбауэровской спектроскопии, рентгеновской и рентгеноэлектронной спектроскопии, масс-спектроскопии, а также электронной микроскопии, мюонного и позитронного методов. Для изучения молекул в адсорбир. состоянии используют флэш-десорбцию (см. Десорбция), все виды оптической и резонансной спектроскопии, дифракцию медленных электронов, магн. методы, методы электронного или ионного проекторов, а также всевозможные электрохим. методы.
А. играет важную роль во мн. природных процессах, в первую очередь в обогащении почв и образовании вторичных рудных месторождений. Явление А. широко используется для разделения сложных газовых и жидких смесей (хроматография), а также смесей электролитов (ионообменная хроматография), в процессах крашения и протравливания, флотации и стабилизации дисперсных систем. А. имеет важное значение в гетерогенно-каталитич. хим. реакциях, во мн. биол. процессах — одним словом везде, где существ, роль играют поверхностные явления.
Лит.: Трепнел Б., Хемосорбция, пер. с англ., М., 1958; Грег С., Синг К., Адсорбция, удельная поверхность, пористость, пер. с англ., 2 изд., М., 1984; Межфазовая граница газ — твердое тело, пер. с англ., М., 1970; Основные проблемы теории физической адсорбции, М., 1970; Адсорбция растворенных веществ, К., 1977; Адамсон А., Физическая химия поверхностей, пер. с англ., М., 1979.
Источники:
- Физическая энциклопедия/Гл. ред. А. М. Прохоров. Ред. кол. Д. М. Алексеев, А. М. Балдин, А. М. Бонч-Бруевич, А. С. Боровик-Романов и др.- М.: Сов. энциклопедия. Т.I. Ааронова - Бома эффект - Длинные линии. 1988. 704 с., ил.
|
ПОИСК:
|
При копировании материалов проекта обязательно ставить ссылку на страницу источник:
http://physiclib.ru/ 'Библиотека по физике'