Сэндвич или волчок?
Структурно-химических появления
Одной из наиболее распространенных структурно-химических причин появления "лишних", т. е. не оправданных предполагаемой структурой, линий в колебательных спектрах молекул служит поворотная изомерия. Знатоки этого явления исходят обычно из твердого убеждения: все, что может повернуться, повернется. И оно обычно так и поступает - поворачивается и стимулирует возникновение дополнительных, то бишь "лишних", линий.
Модельными объектами для изучения законов поворотной изомерии служат обычно предельные углеводороды, в особенности их галоидопроизводные. И все уже привыкли к тому, что обычно богатый спектр газа или жидкости сильно упрощается при переходе к твердой фазе. "Лишние" линии при этом вымораживаются, и вы с удовлетворением рассматриваете спектр одного из индивидуальных конформеров. Какого именно? Ну, тут вступают в действие строгие законы спектроскопии, позволяющие определить симметрию конформера, например судить о том, есть у него центр инверсии или нет... И так далее.
Это все у нормальных углеводородов, у которых нет кратных связей. Ну, а если в молекуле есть кратные связи, и не одна, а сразу две, да еще и в сопряженном положении, тогда как?
В монографиях начала шестидесятых годов можно прочитать, что это случай трудный. Поскольку ординарная связь, находящаяся между двумя двойными, не совсем вроде бы и ординарная, короче говоря, сопротивляется она повороту не хуже двойной и все тут. И наблюдать у диенов переход одной конформации в Другую - дело хлопотное. Но у автора вскоре это получилось, причем в полном соответствии с поведением модельных предельных - в жидкости смесь конформеров, в кристалле - один. По пока еще не на самом простом из диенов. Ведь самый простой из сопряженных диенов - бутадиен - устроен так, что одна из конформаций у него, а именно транс-конформция с плоским расположением сопряженных двойных связей, существенно более выгодна по стерическим факторам, чем вторая плоская, т. е. цис. Во второй существенно взаимное отталкивание близко подошедших друг к другу несвязанных атомов водорода. Можно, конечно, выйти из плоскости, но ведь это явная потеря энергии сопряжения. И в результате молекулы в цис-форме, как считают многие, у бутадиена составляют всего около процента от общего числа и по спектру увидеть их не так легко. Но можно ведь, варьируя заместители, подобрать такой диен, в котором существенные стерические затруднения будут в плоской транс-форме, Тогда относительная роль другой плоской конформаций, т. е. цис, может существенно возрасти, можно даже сделать ее превалирующей.
Здоровая идея "прошла" на 2, 4-диметилпентадиене-1, 3. Здесь как раз в характеристической области колебаний С=С выше 1600 см-1 обнаружилось четыре линии. Связей С=С в диене две, а линии четыре. Значит, две из четырех - "лишние". Понижаем температуру - две из четырех постепенно гаснут. Замораживаем жидкость - те же две полностью уходят из спектра КРС.
Но как в подобном случае разобраться с конформациями, какая из них выморозилась, а какая осталась? Симметрия самой молекулы низкая, центра инверсии нет ни в одной из конформаций, так что на альтернативный запрет рассчитывать нечего. Как же быть?
Выход был найден при исследовании колебательных спектров модельных диенов, т. е. систем с фиксированным расположением двойных связей как транс, так и цис. Оказалось, что существует достаточно строгое правило* согласно которому частота симметричного (а в молекулах с низкой симметрией - синфазного) колебания С=С связей в транс-форме выше, чем антисимметричного (антифазного). А в цис-форме все наоборот!
* (На XX Всесоюзном съезде по спектроскопии это правило, проверенное и подтвержденное еще на десятках дополнительных примеров, называли правилом Соболева - Алексаняна (Киев, сентябрь 1988 г.))
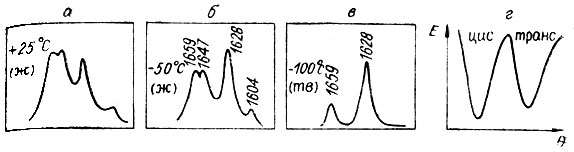
Рис. 26. Нага диеновый сюжет относительно прост, более того, почти классичен. Действительно, в спектрах КРС диена в области колебаний С=С вместо положенных двух линий видим четыре (а), т. е. две из четырех -'лишние'. Снижаем температуру от +25 до -50 °С (б), и интенсивность двух линий из четырех падает. Наконец, замораживаем жидкость, и те же две линии полностью уходят из спектра, как говорят, вымораживаются (в). Все, что происходит со спектрами, прекрасно согласуется с рисунком г, на котором приведена кривая потенциальной энергии диена в зависимости от угла поворота вокруг ординарной С-С связи. А строение двух конформеров, соответствующих минимумам на этой кривой, показано на плакате из рис. 31, III.1
Теперь уже полегче. Ведь отнесение колебаний к синфазному и антифазному по спектрам КРС труда уже не составляет. У синфазного всегда выше интенсивность и ниже степень деполяризации. По этим критериям у нас осталась только цис-форма. Транс-конформация, присутствовавшая в жидкости в сравнимом содержании, выморозилась.
Критерий обращения порядка частот в силу его новизны и неожиданности необходимо было подкрепить и другими. Что же, и степени деполяризации линий С-С в фиксированных транс- и цис-формах оказались различными, что, кстати, нашло и строгую расчетную интерпретацию,- и по этому критерию прошла цис-форма. И ультрафиолетовые спектры поглощения модельных диенов пошли в ход с ослаблением экстинкции и батохромным смещением максимума в цис-конфигурациях - и по этому критерию прошла цис-форма. Так что не так уж и сильно сопротивляется ординарная С=С связь в сопряженных диенах повороту; может быть, и ничуть не сильнее, чем в углеводородах предельных.
Конечно, конечно, но вот если бы бутадиен повернуть и там частоты поменялись бы местами...
С бутадиеном пока немного подождем. Да и вообще хватит нам об органике, вон по ней сколько монографий написано. А вот повернуть бы нечто металлоорганическое, даже ближе к неорганике. Взять хотя бы сэндвич, в коем две дикарболлидные клетки зажали между собой атом металла. Крутанем одну клетку, а вдруг будет волчок?
Ну что же, поговорим о сэндвичах. Методы колебательной спектроскопии с их исключительной структурной чувствительностью нередко вселяют в специалиста самоуверенность. Даже с оттенком снобизма: мол, нет проблем!
Что проблем нет - это неверно. Проблемы, конечно, есть. Здесь намного сложнее, чем в органитке, идентификация характеристических колебаний, а без нее как выйдешь на "лишние"? Те самые "липшие", с которыми надо изрядно повозиться, прежде чем что-либо начнет получаться. Да и исходное состояние здесь чаще всего не газ и не жидкость, а кристалл, а в кристалле ведь - все привыкли - один конформер.
Но почему, собственно говоря, один, обязательно один? Что, может, указ такой вышел - быть в кристалле ему одному, норма, мол, жизни у кристалла такая, и баста. Или существуют какие-либо принципиальные запреты на пару? Природа ведь удивительно разнообразна, так почехму бы ей время от времени не спаривать .два разных конформера в одной элементарной ячейке? Ну, хотя бы для удовольствия спектроскопистов.
Возьмем ряд соединений, устроенных одинаково,- дикарболлидные клетки, а между ними атом металла. Будем менять один металл на другой, немного поиграем и с клетками. И будем смотреть на спектры.
В подавляющем большинстве таких дикарболлидов в низкочастотной области спектра есть одна сильно поляризованная (в растворах) очень интенсивная линия. В спектрах самих дикарболлидных клеток ничего подобного нет, переход от одного металла к другому смещает частоту. В общем, никуда тут не денешься, да и деваться некуда. И все, кто с такими сэндвичами работал, в один голос утверждают: интенсивная линия в спектрах КРС - это линия симметричного валентного колебания металл - дикарболлиды. А справедливость подобного утверждения в шкале вероятностей может быть уценена высшим баллом - 100 %. И оценена без всякой боязни.
Вот одна интенсивная (и очень!) линия, в другом веществе опять одна. Но почему в 1, 7-дикарболлиде никеля (IV) вместо одной очень интенсивной линии - сразу три (?!).
В соответствии с логикой ряда мы теперь можем утверждать, что и вторая, и третья интенсивные линии в спектрах КРС этого соединения - "лишние".
Забегая несколько вперед, заявим сразу, что про третью "лишнюю" мы будем говорить отдельно. Потому что она настолько сильно удалена по частоте (более чем в два раза) от линии основной, вписывающейся в большой ряд, что всякие разговоры о поворотной изомерии как о возможной причине ее появления можно прекращать сразу, даже не начиная. А буде кто-нибудь начнет - пресекать в корне. Тем более, что и степень деполяризации у нее для очень интенсивных линий аномально высока (0,6), да и название для нового очерка уже готово - ТРЕТЬЯ "ЛИШНЯЯ". Согласитесь, что название звучит. А вот про ""лишнюю" вторую, ту самую, что отошла от основной всего на какой-то десяток обратных сантиметров, так что даже и не отличишь, какая из двух в дублете "лишняя", а какая нет, поговорить в самый раз именно теперь. Постойте-ка, а что там по ее поводу писали другие авторы, ведь молекула известна уже по крайней мере с десяток лет?
А другие авторы писали так. Резонансный, писали, это, мол, эффект. Разве вы не знаете, что в кристаллах бывает резонанс Давыдова?
А наше мнение такое - на давыдовское расщепление это не похоже. Потому что оно, как правило, не превышает долей процента от значения частот. А тут сразу более пяти процентов. Нет, не похоже!
А что же тогда? Ведь у нас кристалл, а в кристаллах, как мы знаем, поворотный изомер обычно один. Растворим вещество - дублет исчезает, остается одна низкочастотная компонента. Нет, определенно это эффект Давыдова, ведь он специфичен исключительно для кристаллов.
А вот не эффект Давыдова и все. Не Давыдова! И чтобы доказать сие утверждение, надо нам придумать нечто такое, такое... ну в общем такое, чего еще не было. Какие там тесты с поворотными изомерами проходили? Замораживание? Что же, попробуем для начала наш раствор заморозить и снимем спектр.
Но в спектре опять пара линий, точно такая же, как в кристаллической фазе. При этом даже невооруженным глазом видно, что бензольный раствор, замерзая, расслаивается, выталкивает из себя всю желтизну нашего дикарболлида, т. е. выпадает из него кристаллическая фаза и ведет себя точно так, как ей и положено себя вести.
А если наш раствор заморозить побыстрее? Очень быстро. Так быстро, чтобы не успела произойти кристаллизация, чтобы не успели вытолкнуться из застывающего растворителя молекулы дикарболлида, чтобы не успели объединиться в своей фазе. Что, трудно такой эксперимент провести? А алмазная кювета на что?
Времени на подготовку и проведение эксперимента ушло не так много, всего несколько дней. Но зато результат... Да, результат получился впечатляющий. Низкочастотная линия дублета, практически доминирующая в бензольном растворе, полностью перекачалась всей своей интенсивностью в линию высокочастотную. То есть один поворотный конформер полностью перешел во второй!
Удачный эксперимент подогрел страсти. Что бы такое еще совершить, не менее, а может быть, и более оригинальное? Такое, чего еще никогда не было?
Помог случай. Захотелось посмотреть, какие изменения в спектр внесет переход к другой возбуждающей линии, ну, скажем, от красной к зеленой. А зеленая-то линия помощнее, да и поглощается в нашем ярко-желтом веществе сильнее. И, невзирая на эффективный теплоотвод в алмазной кювете, начал кристаллический порошок от зеленого света слегка подгорать. И спектр соответственно стал слегка портиться, все интенсивности начали снижаться. Но что это? Одна из линий дублета, низкочастотная, выжигается с большей скоростью. Вот она почти полностью выгорела. А высокочастотная еще в силе!
Так родился второй тестовый новый метод работы с "лишними" линиями, метод фотовыжигания. С точки зрения поворотной изомерии он вполне оправдан, ведь хорошо известно, что реакционная способность разных конформеров различна. Взять хотя бы те же диены - в знаменитую реакцию диеновой конденсации вступает только цис-конформация. Да что там диены, даже у белков эта разница известна, читайте соответствующие монографии,
Дальше - больше. Захотелось попробовать, нельзя ли применить в конформационном анализе пористые стекла. А что, введем в мельчайшие поры размером около 80 Å наш раствор, запишем спектр и...
...Низкочастотная линия перешла в высокочастотную. Опять молекула повернулась?!
Попять сей феномен можно по-разному. Но автору очень хочется объяснить его следующим образом. В порах, по его мнению, достаточно высокие градиенты электростатического поля. И это поле разворачивает молекулы так, чтобы результирующая конформация давала наибольший энергетический выигрыш - ну хотя бы за счет взаимодействия поля с дипольным моментом молекулы.
Почему манит именно такая интерпретация? Да потому что... если повернуть из трапс- в цис-конформа цию бутадиен, то у него появится дипольный момент. А ведь очень хочется его повернуть и проверить наконец, работает ли для него, простейшего, это эмпирическое правило обращения частот, когда vs становится ниже vas.
Эксперименты с бутадиеном - впереди. Ну а как поживает наша третья "лишняя"? Как реагирует она на все описанные выше операции с дикарболлидом?
В растворе с низкочастотным изомером она остается, при фотовыжигании низкочастотного изомера тоже. Значит, принадлежит она одинаково как тому, так и другому изомеру.
Так что же это, собственно говоря, такое, третья "лишняя"? Линия, которой не должно быть, но которая есть, и наплевать ей на взаимный разворот дикарболлидных клеток.
Автор, взирая на третью "лишнюю", почему-то сразу вспомнил рассказ своего учителя по спектрохимии профессора Вилена Тачатовича Алексаняна. Вилен Тачатович в конце семидесятых побывал в Канаде и по приезде рассказал об оригинальных работах канадского ученого Кеннингстайна, который...
Впрочем, и о Кеннингстайне, и о любопытнейшем поведении третьей "лишней" в неких особых обстоятельствах у нас разговор отдельный.
|
ПОИСК:
|
При копировании материалов проекта обязательно ставить ссылку на страницу источник:
http://physiclib.ru/ 'Библиотека по физике'